How to Read an Ssr Pedigree Tree
Introduction
Uncomplicated sequence repeats (SSR) – otherwise known as microsatellites – exist ubiquitously throughout prokaryotic and eukaryotic genomes (Tóth et al., 2000). Based on their universal distribution and loftier density in a multitude of genomes, SSRs have been analyzed as second-generation molecular markers. Given their high rates of mutation, SSRs are widely used in genetic analysis, factor mapping, quantitative trait locus (QTL) mapping, and marker-assisted selection (MAS) breeding. SSRs in DNA coding regions are used as anchor markers for specific populations due to their homology amongst related species, while the large variations in SSRs found in non-coding regions provide adequate polymorphisms to distinguish related species. Hence, SSR markers have been specifically applied in a variety of identification procedures, allowing for the successful structure of a DNA fingerprinting database that includes the cultivars of a number of crops, such every bit maize, wheat, and watermelon (Zhang et al., 2012; Tian et al., 2015; Wang et al., 2015, 2017). Even so, many SSRs used in previous studies were often less polymorphic and failed to yield the expected PCR products. This limited the utilize and accuracy of SSR markers for genotyping in genetic research (Gao et al., 2012; Hu et al., 2014; Li et al., 2017).
Traditional gel electrophoresis cannot distinguish base differences or changes correctly in SSR amplicons, often causing false positive or false negative results in SSR detection, likely caused past sequence variations in the SSR motifs or their flanking sequences; these variations may touch the PCR procedure and hence, the resultant products. Recently, genome-broad analyses of SNPs, SVs, and transposon insertion polymorphisms (1 of several transposable elements or TEs) were conducted based on large-scale resequencing studies in genetic variome research (Qi et al., 2013; Yang et al., 2017). Still, few studies have focused on genome-wide SSRs, especially perfect SSRs, which exhibit stable motifs and conserved corresponding flanking sequences. To appointment, few studies have attempted to characterize genome-wide perfect SSRs. The few that do exist in the literature have focused on the SSR motifs themselves without looking farther into their flanking sequences (Ding et al., 2017; Yasodha et al., 2018). Therefore, the identification of genome-wide perfect SSRs with stable motifs and corresponding flanking sequences that are highly conserved is disquisitional in crops, such that amplification of the appropriate PCR products can exist ensured in genetic inquiry applications. Information technology volition remain incommunicable for the enquiry community to attain this goal without admission to a loftier throughput technology for SSR genotyping.
A recent study established Ampli-seq as the commencement high throughput SSR genotyping method based on second-generation sequencing technology utilizing the Illumina MiSeq platform (Li et al., 2017). This report reported that the toll for the capture and detection of multiple SSRs in each rice line was $40 and $5, respectively; an boilerplate of 2427.75 SSRs was obtained out of a total of 3105 SSR targets in viii rice lines, with SSR coverage of 1855.38 and a genotyping success rate of 78.19% (Li et al., 2017). Even so, the rapid development of high throughput sequencing engineering has yielded several novel and more economical sequencing platforms, such as the Illumina HiSeq X Ten (Ten Ten) and the NovaSeq (Meynert et al., 2014; Costello et al., 2018). These instruments provide opportunities to develop novel high throughput SSR genotyping technologies when combined with genome-wide perfect SSR discovery at a lower toll and with higher success rates than currently available methods.
In this study, we developed a novel method called Target SSR-seq, which combined the high throughput sequencing organization X 10 platform with genome-wide perfect SSRs that harbored stable motifs and flanking sequences derived from 182 resequencing datasets of a cadre collection of cucumber lines. This method enables the genotyping of hundreds of targeted SSR loci in a big number of samples with loftier coverage, simultaneously in a single Illumina HiSeq lane (Yang et al., 2016). By calculation sequencing adapters and dual barcode tags (Campbell et al., 2015), the SSR genotypes were adamant directly from the deep sequencing (∼1000×) of PCR products. The present study constructed the DNA fingerprints of 382 cucumber varieties with 89 genome-broad perfect SSRs and 22 well-known SSRs for varieties identification using the Target SSR-seq engineering. The assay required 72 h for high throughput genotyping at a cost of $vii for each variety, demonstrating the high utility of this new approach. This written report developed a core set of perfect SSRs in cucumber, including backbone varieties, which demonstrated their breeding history in Communist china.
Materials and Methods
Plant Materials and Dna Extraction
A total of 382 commercial cucumber varieties were utilized in this study (Supplementary Table S1), including 115 varieties from the seed section of the Chinese authorities, 146 varieties from breeders, 91 commercial hybrid varieties from seed markets, and 31 varieties cultivated in Xishuangbanna. Offset truthful leaves from 30 independent individuals, which was required based on the National Varieties Identification Standard, were collected and mixed to extract DNA following a CTAB-based method in this study (Stewart and Via, 1993).
Discovery of Genome-Broad Perfect SSRs in Cucumber
First, the cucumber reference genome 9930 V2 was analyzed to uncover genome-wide SSRs using GMATA software with the post-obit parameters: motif repeated at least three times, motif length at least 3 bp, and repeat length upward to 100 bp (Huang et al., 2009; Wang and Le, 2016). In order to select the suitable SSR loci for Target SSR-seq, we extracted SSRs with 2 bp motif repeats at least six times, 3 bp motif repeats at least five times, 4 bp motif repeats at to the lowest degree 4 times, 5 bp motif repeats at to the lowest degree three times, and vi bp motif repeats at least two times. Moreover, 15-bp flanking sequences of SSR loci on the reference genome were mapped to reference genome using BWA, and SSRs with unique matches were retained. Second, a collection of resequencing data from 182 genetically diverse cucumber accessions, including the 115 published lines (SRA056480 in NCBI, Qi et al., 2013) as well equally the 67 unpublished resequencing data (Supplementary Table S2), was used to discover genome-wide perfect SSRs. The perfect SSRs were constrained using the following criteria: (i) SSR motif length less than 50 bp; (ii) no INDELs, poly regions, and SSR loci in the 150 bp flanking sequence; (iii) read frequency of the major SSR allele in one accretion greater than 0.seven to reduce the noise when BWA immune mismatch; (iv) Picture show value greater than 0.3 to ensure the SSR polymorphism in varieties. (five) Even distribution in chromosomes. Finally, a multiplexed PCR panel of the selected perfect SSRs was designed by Molbreeding Biotechnology Company (Shijiazhuang, China).
In addition, 58 well-known SSR loci used to distinguish cucumber varieties in Cathay (NY/T 2474-2013) (Lv et al., 2012) were analyzed based on the criteria for multiplexed PCR, 31 of which were retained to compare genotyping efficiency to that in the perfect SSRs.
Target SSR-Seq Library Construction
The Target SSR-seq library construction consisted of ii rounds of PCR (Figure one): the showtime circular amplified and captured the target SSRs in found Deoxyribonucleic acid samples using a multiplexed PCR panel; the second round added a unique barcode to the capture product for each Deoxyribonucleic acid sample. First, the multiplexed PCR was conducted in 30 μL reactions including fifty ng DNA template, 10 μL of 3 One thousand enzymes, and 8 μL of the multiplexed SSR-capture panel mix (Molbreeding Biotechnology Visitor, Shijiazhuang, China). The PCR weather were equally follows: 95°C for 5 min and then 17 cycles of 95°C for thirty southward and 60°C for four min, and extension at 72°C for iv min. The PCR products were purified past magnetic dewdrop suspension and fourscore% booze. And so the second PCR was performed in 30 μL reactions consisting of 11 μL of purified PCR product from the previous circular, 10 μL of 3 M Taq enzyme, eighteen μL pure water, and ane μL of primers with the post-obit sequences: forrad 5′-AATGATACGGCGACCA-CCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCG-iii′ and contrary 5′-CAAGCAGAAGACGGCATACGAGATXXXX XXXXGTGACTGGAGTTCCTTGGCACCCGAGA-iii′ (barcodes are indicated by underlined sequences). The PCR atmospheric condition were as follows: 95°C for three min so 7 cycles of 95°C for 15 s, 58°C for fifteen s, and 72°C for thirty s, and extension at 72°C for 4 min. The second round PCR products were purified with 100 μL 80% alcohol and 23 μL Tris–HCl buffer (x mM, pH eight.0–viii.five). Thereafter, the Target SSR-seq library was ready to sequence on the X Ten platform (Molbreeding Biotechnology Company). To verify the repeatability of Target SSR-seq, DNA of cucumber 9930 and pure water were gear up as positive and negative controls in PCR amplification, respectively.

Figure 1. Target SSR-seq pipeline. Schematic workflow of perfect SSR choice, Multiplexed PCR pattern, high-throughput sequencing, and accurate SSR genotype.
SSR Genotyping Analysis of Target SSR-Seq
The raw data reported in this report have been deposited in the Genome Sequence Archiveone under accession numbers CRA001490. The raw Target SSR-seq data were de-multiplexed to make up one's mind the exact genotypes for each diverseness using the Illumina bcl2fastq pipeline (Illumina, San Diego, CA, Usa). Adaptor and low-quality sequences were filtered out from raw reads using Trimmomatic with parameters as "SLIDINGWINDOW: iv:20 LEADING:3 Abaft:three MINLEN:twoscore" (Bolger et al., 2014). The reads of each variety were mapped to the cucumber reference genome (9930 V2)ii using BWA with default parameters and 15-bp flanking sequences of SSR loci on reference genome were isolated to determine the perfect SSR genotype using MISA softwarethree (Li and Durbin, 2009). Based on the high-throughput sequencing results, the SSR alleles with the maximum numbers of reads and the second maximum numbers of reads were treated equally the major and minor allele for each target SSR loci. When the read frequency of the major allele was more than 0.7, this locus was described every bit homozygous. When the read frequencies of the major and minor alleles were both more than 0.35, this locus was treated as heterozygous.
Genetic Data Statistics for Target SSRs
Genetic information statistics including SSR allele number per locus, observed heterozygosity (Ho ), genetic diversity, polymorphic information content (Moving-picture show) value (Botstein et al., 1980), and inbreeding coefficient (F, Wright, 1965) were calculated using a Perl script with the post-obit equation:
where l is the allele locus and Pi and Pj represent the population frequency of the ith and jth allele.
Genetic Structure Analysis in Cucumber Varieties
Population structure was inferred past a model-based program STRUCTURE V2.3 with the following parameters: 100,000 burn-in length, ten,000 iterations, admixture model (Pritchard et al., 2000; Falush et al., 2003). The optimal number of ancestors (K) was determined using the ΔK method with Chiliad ranging from i to 10. The population of individuals was defined by the proportional membership. Furthermore, a "hierarchical Construction assay" was practical to suspect the potential subpopulation structure (Vähä et al., 2007; Emanuelli et al., 2013). A hierarchical clustering on main components (HCPC) analysis was performed to validate the results divers by STRUCTURE, with HCPC office in the FactoMineR R package (Lê et al., 2008; Husson et al., 2014). The variance between clusters, variance gain, and variance ratio were calculated with the cluster number Q ranging from 1 to 10. The optimal cluster was determined by the minimized variance ratio. In addition, a principal co-ordinate assay (PCoA) and an unrooted Neighbour-joining tree with Nei's standard genetic distance were performed using the ape and poppr packages in R software (Nei, 1978; Kamvar et al., 2014).
Population Differentiation Analysis in Cucumber Varieties
To measure genetic differentiation between populations, we performed an analysis of molecular variance (AMOVA); the pairwise Fst. AMOVA was performed in the poppr R package (Kamvar et al., 2014) and the pairwise Fst was performed with the hierfstat R package.
Cadre SSRs Set Exploration for Varieties Identification
To select a core SSR prepare for diversity identification, we developed a new Perl method to cull the best discernibility group based on the principle of minimum numbers of SSRs representing the maximum genetic diversity. Discernibility by pairwise comparison of all samples was as the first filter status, and the dataset with the same discernibility were and so selected with higher Movie. The highest discernible SSR loci were called as an initial cadre dataset and each SSR were later on added to the initial core dataset to course a new dataset. The second SSR were called from the new dataset with highest discernibility and were added to the core dataset. The following choice were the same as the second SSR until the discernibility reached the maximum. Finally, a best-discernibility group of SSRs was obtained as the core SSR set, and the saturation curves of its discernibility were plotted by pairwise comparison of varieties genotypes.
Core Varieties Analysis in Chinese Cucumber Markets
According to the international standards for identifying crop varieties (International Union for the Protection of New Varieties of Plants [UPOV], 2011), we fix a pairwise comparing matrix past calculating the numbers of differential SSR genotypes between each variety and the remaining ones; the missing genotype was treated equally aught. Fewer differential SSR genotypes indicated closer kinship with others. The top 10% of varieties with close kinship were considered core varieties in each group.
Results
The Novel Target SSR-Seq Pipeline
In this study, we adult a novel approach for SSR genotyping using a target sequencing technology called Target SSR-seq, which can be applied in genetic research, Dna fingerprinting, variety identification, and molecular breeding (Figure 1). This study tested the Target SSR-seq pipeline in cucumber, the genome of which is well assembled (Huang et al., 2009; Li et al., 2011). Start, we selected the candidate SSR loci to exist genotyped. Second, nosotros designed a multiplexed PCR procedure to capture target SSR regions in a plant genome. Then the Target SSR-seq library was sequenced on the loftier-throughput sequencing platform X Ten; each SSR region was sequenced for at least one thousand× coverage. To assay the repeatability of Target SSR-seq, positive and negative controls of PCR amplification were fix. The cucumber 9930 was ready as a positive control, and the pure water was used as a negative control in the Target SSR-seq experiment. The amplification and sequencing result showed that the genotype of 91 perfect SSRs was the same to that in the cucumber reference genome (9930 V2). While equally the negative control showed no PCR bands later screening in agarose electrophoresis. The positive and negative controls proved that Target SSR-seq could obtain reliable genotyping result.
Compared with existing methods for SSR detection, Target SSR-seq combined multiplexed PCR with target deep sequencing and was immediately capable of highly accurate SSR genotyping (Supplementary Tabular array S3). This new engineering successfully genotyped hundreds of target SSR loci in numerous samples within 72 h at a toll of $seven for each variety, which was more efficient and cost-effective than the previously reported Amp-Seq SSR technology (Supplementary Tabular array S3).
Discovery of Genome-Wide Perfect SSRs for Target SSR-Seq
We acquired 208 139 SSRs in the cucumber reference 9930 V2 genome, of which 10 404 SSRs were suitable for multiplexed PCR capture. Based on the resequencing data for 182 cucumber varieties, 1700 SSRs exhibited polymorphisms. Furthermore, 844 perfect SSRs were obtained with read frequency of the major alleles greater than 0.7 and stable flanking sequences. In this study, 91 evenly distributed perfect SSRs in the cucumber genome were randomly selected to test in Target SSR-seq.
In addition, the current Target SSR-seq panel included 31 SSR loci that are oft tested in genetic research on cucumbers, which were used to compare the genotyping efficiency with that of the genome-wide perfect SSRs. Finally, a total of 122 target SSRs were successfully designed in the next multiplexed PCR procedure.
Genotyping Analysis in Target SSR-Seq
In total, the Target SSR-seq obtained 230 1000000 reads and 34 billion bases in 382 cucumber varieties (Figure 2). In the 122 target SSR loci, six SSRs from the 31 compared SSRs failed to be genotyped due to low motif capture (80.six% success rate), while the 91 tested perfect SSRs were successfully genotyped (100%). The average coverage of the 116 retained SSRs in each sample was 1289× (Figures 2, 3A). Among the 382 varieties, 375 varieties (98%) showed more than 90% alignment rate to the 9930 V2 reference genome (Figure 2A). Out of these aligned reads, 372 varieties (97.4%) exhibited an alignment rate to the target SSR motif over 98%, and the alignment rate in all 382 varieties was above 95% (Figure 2B). The average read depth per SSR capture in 311 varieties (81.5%) was more 1000× (Figure 2C). Furthermore, we analyzed the Target SSR-seq uniformity alphabetize, in order to calculate the proportion of the coverage above 10% of mean depth value for each variety (Nishio et al., 2015). The average uniformity index in this study was 89.5% (Figure 2nd), indicating a higher uniformity alphabetize and more accurate results. Moreover, 2 SSRs from the 25 retained for comparing harbored no polymorphisms in 382 varieties, and i SSR exhibited a high miss rate (>20%), probably due to an unstable flanking sequence. 2 of the 91 perfect SSRs were observed as monomorphisms in 382 varieties. Finally, nosotros obtained 111 polymorphic SSRs for genotyping 382 cucumber varieties from Chinese markets.
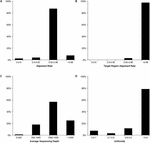
Figure two. Target SSR-seq genotyping result analysis. The distribution of reads alignment (A), target region alignment (B), average read depths (C), and uniformity alphabetize (D) for 382 cucumber varieties.
Genetic Diversity of Cucumber Varieties in China
Target SSR-seq captured 398 alleles of 111 target SSR loci in 382 varieties and the allele number per SSR locus varied from 2 to 12 with an boilerplate of 3.6 (Figure 3B). Trinucleotides and dinucleotides were the commencement two motif types, bookkeeping for 37.4 and 28.8% in 398 alleles, respectively (Supplementary Figure S1A). The SSR motif repeats ranged from ii to 23, and 127 alleles (32%) independent two repeat units (Supplementary Figure S1B). There were 239 alleles (60%) with minor allele frequency (MAF) above v% that were regarded as mutual alleles, while only 20.iii% were plant in a previous report (Lv et al., 2012). In the 159 rare alleles, 28 (17.5%) were specifically observed in Xishuangbanna varieties. We constitute the observed heterozygosity Ho varied from 0 to 0.95 with a mean of 0.17, and seven SSRs exhibited higher Ho (>0.4) (Figure 3C). The depression Ho indicated a narrow genetic background in 382 Chinese cucumber varieties. Furthermore, the genetic diverseness estimated by expected heterozygosity varied from 0.003 to 0.809 (mean = 0.367, Figure 3D), while the Moving-picture show value ranged from 0.003 to 0.782 (hateful = 0.310, Figure 3E). Interestingly, the inbreeding coefficient of four perfect SSR loci was negative, indicating that these loci had excess heterozygosity. Overall, the 111 target SSR loci showed various alleles and loftier polymorphism rates, which were proven to be suitable for varieties identification.
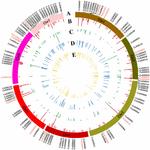
Figure iii. Genetic characterization of 111 SSRs in 382 cucumber varieties. (A) Distribution of 111 SSR loci in seven cucumber chromosomes. 16 core SSR set is labeled in red. (B) Allele numbers per SSR locus. (C) Observed heterozygosity. (D) Genetic diversity. (E) Movie value.
Genetic Construction of Cucumber Varieties in Cathay
The STRUCTURE and Evanno's correction results indicated that 382 cucumber varieties were divided into two main populations (Pop1 and Pop2), based on the optimal number of K = ii (Figures 4A,B). In general, 276 cucumber varieties (72.1%) were assigned to Pop1 and the remaining 107 varieties were assigned to Pop2. To find the subpopulation construction, a hierarchical STRUCTURE analysis was performed. The Pop1 was divided into Pop1A and Pop1B, while the Pop2 was equanimous of Pop2A and Pop2B (Figure 4C). A total of 99% partition defined by "hierarchical Construction analysis" was the same as those retrieved from the outset round Construction analysis when Thou = 4, which agreed with the plateau criterion proposed by Pritchard et al. (2010). According to its geographic origin, Pop1A belonged to northern Mainland china cucumber, Pop1B indicated the southern Prc cucumber, while Pop2A represented cucumber derived from Europe and Pop2B inferred the unique Xishuangbanna cucumber.
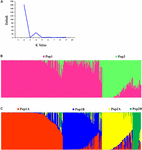
Figure 4. Population structure of 382 cucumber varieties. (A) Delta Chiliad plots derived from Target SSR-seq consequence. (B) Two populations were observed in 382 varieties, Pop1 is colored in pinkish and Pop2 is colored in green. (C) Four subpopulations were classified and Pop1A, Pop1B, Pop2A, and Pop2B are colored with red, bluish, yellowish, and greenish, respectively.
HCPC assay was used to validate the results from Construction. The variance between cluster and the variance gain were significantly decreased when the cluster number increased (Supplementary Figure S2). The recommended two clusters inferred by the minimum variance ratio was consistent with analysis on Structure. However, the variance gain increased slowly with cluster numbers across four (Supplementary Figure S2), indicating that four singled-out sub-clusters existed. Furthermore, a hierarchical clustering tree as well demonstrated two clusters and four sub-clusters (Supplementary Figures S3, S4). Moreover, the unrooted Neighbor-joining (NJ) tree and Principal co-ordinates assay (PCoA) indicated a clear distinction in ii populations and 4 subpopulations, despite the fact that Pop1B was shut to Pop1A (Figures v, 6). Effigy 5 and Supplementary Figure S3 likewise showed that Pop2A were divided into two branches, one was typical European fruit types, and the other one was European fruit types which interbreed with southern China cucumber.
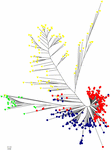
Figure 5. Unrooted neighbor-joining tree of 382 cucumber varieties. The Pop1A, Pop1B, Pop2A, and Pop2B subgroups are colored the same as in Figure 4.
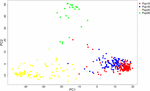
Figure half-dozen. Principal co-ordinates analysis (PCoA) of 382 cucumber varieties. The Pop1 and Pop2 are labeled with circle and triangle. The 4 subpopulations are colored the same as in Effigy 4.
Population Differentiation of Cucumber Varieties in China
AMOVA analysis of 111 SSR genotypes in 382 varieties indicated that the maximum variation of 29.two% resulted from differences within samples, while the minimum variation of 17.6% was accounted for betwixt subpopulations within populations (Table one). The Fst consequence demonstrated that population differentiation between Pop1 and Pop2 is moderate (Fst = 0.35), which was similar to previous research in cucumber germplasms, ranging from 0.xxx to 0.33 based on 23 SSRs (Lv et al., 2012). The pairwise Fst between four subpopulations ranged from 0.14 to 0.47 (Table 2). Amongst them, the Fst between Pop1A and Pop1B showed a low level of differentiation (Fst = 0.fourteen). The distinct differentiation was observed in other pairwise Fst analysis.

Tabular array 1. Analysis of molecular variance (AMOVA) among populations and inside populations.
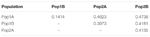
Table two. Pairwise differentiation Fst among four subpopulations.
Core SSR Fix in Cucumber Varieties Identification
The core SSR set was used to analyze the genetic diversity and diverseness identity in crops (Lv et al., 2012; Zhang et al., 2016). This report constitute that a set of xvi cadre SSRs could distinguish 99% of 382 commercial cucumber varieties (Figures 2A, 7 and Supplementary Table S4) and the similar varieties (1%) could be distinguished with two SSRs. Structure analysis based on 16 SSRs classified the 382 varieties into 2 populations (Supplementary Effigy S5). The PCoA analysis significantly distinguished the two populations with PC1 explained by 18.vi% and PC2 explained past 9.four%, respectively (Supplementary Figure S6). The AMOVA analysis showed that the variations were evenly distributed in populations, samples inside populations, and within samples (Supplementary Table S5). The pairwise Fst between Pop1 and Pop2 was 0.25. Hence, this set of sixteen core SSRs was sufficient in representing the genetic variety and identifying cucumber varieties in Chinese markets.
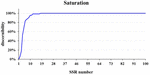
Figure 7. The saturation curve of 111 SSRs identifying in 382 cucumber varieties. A total of 16 SSRs identified 99% cucumber varieties.
Genetic Similarity and Core Varieties Analysis
The genetic background of Chinese cultivated cucumber was considerably narrow, given that breeders follow similar breeding goals, resulting in many varieties with close genetic relationships. In this study, we congenital a genetic similarity matrix in iv subgroups past counting the number of differential SSR genotypes between each DNA sample (Supplementary Effigy S6). Loftier genetic similarity was observed within cucumbers belonging to northern China type, suggesting a long breeding history and all-encompassing gene exchange in this grouping (Supplementary Figure S7A), while the European cucumber blazon exhibited high genetic diverseness, according to its recent introduction into China (Supplementary Figure S7C). Amidst 382 cucumber varieties, "Jinyou1hao" had the minimum number of differential SSR genotypes with others, while a European variety "Virginia" had the maximum differential SSR genotypes. Nosotros selected the top ten% of varieties with minimum differential SSR genotypes as core varieties within each subgroup. Finally, 42 varieties were identified and considered to be core or backbone varieties of 382 cucumber varieties, which was in accordance with breeders' views (Supplementary Table S6).
Word
Loftier Accuracy and Efficiency of Target SSR-Seq
Uncomplicated sequence repeats (SSRs), also known equally brusque tandem repeats (STRs) or microsatellites, exist extensively throughout eukaryotic genomes and are therefore used widely in genetic background pick and MAS breeding, every bit well equally in map-based cloning, QTL mapping, seed identification and purification (Wang et al., 2017). However, few studies have focused on the accurateness and authenticity of SSR genotypes. Due to the high number of variations existing in SSR motifs and flanking regions, the bachelor methods for SSR genotyping often generate faux positive or faux negative results (Li et al., 2017). Therefore, the inquiry community needs development of novel methods for perfect SSR discovery and genotyping that require less time and cost less, while delivering high accuracy and efficiency. In this study, Target SSR-seq genotyped hundreds of perfect SSRs using a high-throughput resequencing method that yielded accurate results due to coverage as high as 1289× (Figure 2C). Moreover, the positive control result showed that the Target SSR-seq of cucumber 9930 obtained the same genotyping results with that in reference genome sequence (9930 V2). And the negative control result showed no PCR amplification. This proved that the Target SSR-seq could proceeds preferable repeatability. Compared to traditional SSR genotyping methods, the efficiency of Target SSR-seq is hundreds of times higher, acquiring dozens to thousands of datapoints in 72 h at a cost less than $vii for each sample. Compared to the recently reported Amp-Seq SSR method (Li et al., 2017), our study gained a genotyping success charge per unit of 100% based on perfect SSRs while 78% was obtained with Amp-Seq SSR; Target SSR-seq also requires less time and fewer consumable materials past utilizing a high-throughput sequencing platform (Effigy one). In addition, the 100% success charge per unit of 91 perfect SSRs was more than the eighty.half dozen% of 31 compared SSRs, commonly used in previous studies (Lv et al., 2012). Therefore, Target SSR-seq succeeds in providing high-throughput SSR genotyping with high accuracy and efficiency for genetic enquiry.
Powerful Awarding in Varieties Identification of Target SSR-Seq
With the development of domestic and international seed trade, the commercial quality of seed based on authenticity and purity is becoming more than important for both seed producers and farmers (Gao et al., 2012). The traditional manner to measure seed authenticity and purity relies on field investigation, which is time-consuming and labor-intensive and unsuited for the fast-paced inspection demands of today (Tian et al., 2015). Recently, UPOV (the International Union for Protection of New Varieties of Plants) proposed SSR markers for variety identification and Dna fingerprinting data base construction (International Union for the Protection of New Varieties of Plants [UPOV], 2011). To date, DNA fingerprinting database using SSR markers was successfully built in cultivars such equally rice, maize, wheat, watermelon, cucumber, and melon. However, the sequence variations of motif and flanking regions in these SSRs were non clearly known, causing a sure amount of SSRs to yield poor results when screened in diverse genetic accessions. Amp-Seq SSR as a new method was able to genotype more than thousands of SSRs at once using high throughput-sequencing technology and was successfully applied in rice research (Li et al., 2017). Moreover, it is user-friendly to apply fewer numbers of SSR markers rather than thousands of markers in identifying varieties, especially in vegetable crops due to pocket-size genomes and limited numbers of varieties in markets. Thus, this study calculated a core set of xvi perfect SSRs to place varieties and ready DNA fingerprints successfully. Consequently, Target SSR-seq combined with perfect SSRs is a powerful method for genetic analysis and varieties identification.
Genetic Diversity Analysis in Chinese Cucumber Varieties
It was well known that Red china has a long history in the cultivation of cucumbers since the Han dynasty, when it has been reported that cucumber was outset introduced into Cathay through the Silk Road (Lv et al., 2012). Over several k years of human selection and improvement, Chinese cucumbers have gained special features (Qi et al., 2013), particularly in fruit length. Over the last 30 years, many modern European varieties and resources were again introduced to Communist china, improving the traditional Chinese cucumber varieties. To date, People's republic of china is the world's meridian producer and consumer of cucumbers, with over one.16 1000000 hectares in cultivated acreage and about 61.9 million tons of product in 20164. However, the genetic background has remained unclear, likewise as the diversity of cucumber varieties in current Chinese markets. This study created a novel method called Target SSR-seq, which successfully genotyped 111 genome-wide SSRs in 382 cucumber varieties in Chinese markets. The results showed four subpopulations were constitute: northern Mainland china type, southern Cathay type, European blazon, and Xishuangbanna blazon (Figure 4C), which was consistent with the geographic distributions (Lv et al., 2012). Withal, including material from Bharat is likely to modify these patterns. In addition, we identified 42 core cucumber varieties by counting the number of differential SSR genotypes of each variety compared to other ones (Supplementary Effigy S7), which was inconsistent with the definition of core resources collection. The core varieties generally harbored more common alleles within groups. Accordingly, the Jingyouyihao diverseness had high genetic similarity with other varieties, and several Europe varieties had high genetic multifariousness compared with other groups. This was in accordance with cucumber breeding history in past decades.
Potential Applications of Target SSR-Seq
In view of its high accuracy and efficiency, Target SSR-seq associate with genome-wide perfect SSRs has cracking potential awarding not just in varieties identification, merely as well in many other inquiry fields (Zhang et al., 2012; Weng et al., 2015; Li et al., 2017), such equally genetic background choice, gene mapping procedures, QTL mapping, and molecular breeding. Furthermore, the Target SSR-seq technology provides a great potential opportunity to apply well-studied SSRs explored by the global enquiry community, in order to ready up a novel molecular design breeding panel. To date, there were dozens of published functional SSRs in cucumber, similar powdery mildew resistance (He et al., 2013), early flowering (Lu et al., 2014), perfect flower (Tan et al., 2015), female flower fourth dimension (Bo et al., 2015), fruit peduncle length (Vocal et al., 2016); parthenocarpy (Lietzow et al., 2016; Wu et al., 2016), downy mildew resistance (Wang et al., 2016), fruit length (Pan et al., 2017), and waterlogging (Xu et al., 2017). Combining these functional SSRs with target SSR-seq applied science, this technology would be practical in a breeding system to greatly raise breeding efficiency and decrease pyramiding convenance flow. In decision, Target SSR-seq tin be widely used in many research fields.
Writer Contributions
CW designed the inquiry. JY did the bioinformatics analysis. CW, RH, FZ, AM, and HT prepared the research. JianZ, JL, BD, and HL performed the research. JiananZ designed the multiple PCR. YJ, JianZ, and RH analyzed the data and wrote the manuscript. All authors read and approved the last manuscript.
Funding
This work was supported in part by grants from National Key Research and Development Program of China (2016YFD0100204, 2017YFD0102004), Beijing Academy of Agriculture and Forestry Sciences (KJCX20170402, KJCX20161503, QNJJ201810, KJCX2017102, and JNKYT201601), National Central Technology R&D Programme of Cathay (2015BAD02B00, 2014BAD01B09), Beijing Municipal Department of System (2016000021223ZK22), Beijing Nova Program (Z181100006218060), Beijing Municipal Science and Technology Commission (D171100002517001), Ministry of Agriculture and Rural Diplomacy, Cathay (11162130109236051).
Disharmonize of Interest Statement
The authors declare that the enquiry was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would similar to give thanks professor Zhang Zhonghua (China Academy of Agricultural Science) and Ren Huazhong (People's republic of china Agricultural University) in supplying parts of high throughput resequencing data, and professor Miao Han (China Academy of Agriculture Science) in providing cucumber varieties.
Supplementary Textile
The Supplementary Cloth for this article tin can be establish online at: https://www.frontiersin.org/manufactures/ten.3389/fpls.2019.00531/total#supplementary-material
FIGURE S1 | The distribution of motif repeats (A) and motif length (B) in 398 alleles for 382 cucumber varieties.
FIGURE S2 | The variance in HCPC analysis. Variance in blueish bars (left ordinate), variance gain (left ordinate) in orange confined and variance ratio in green bars (right ordinate) changed with cluster number.
FIGURE S3 | Hierarchical tree produced by HCPC. Four branches (Pop1A, Pop1B, Pop2A, and Pop2B) were obtained and colored with cherry-red, blue, yellow, and green, respectively.
FIGURE S4 | Master component assay of 382 cucumber varieties by HCPC. Pop1A, Pop1B, Pop2A, and Pop2B are labeled in red, blue, yellow, and green blocks, respectively.
FIGURE S5 | Population structure assay with 16 core SSRs set in identifying cucumber varieties. (A) Delta K plots derived from 16 core SSRs set in 382 varieties. (B) Two observed populations were consistent with results from 111 SSRs.
Figure S6 | PCoA assay with 16 core SSRs set. Pop1 and Pop2 are labeled in pink and light-green, respectively.
FIGURE S7 | Heatmap of pairwise comparison matrix derive from differential SSR genotypes in Pop1A (A), Pop1B (B), Pop2A (C), and Pop2B (D). Crimson to Blue indicated the increasing differential SSR genotypes.
TABLE S1 | Information of 382 cucumber varieties in Chinese market.
Tabular array S2 | Information of 67 unpublished resequencing data used in this written report.
TABLE S3 | Comparisons of Target SSR-seq in SSR genotyping with current methods.
TABLE S4 | The genetic characteristic data of 16 cadre SSRs set up in cucumber diversity identification.
TABLE S5 | Analysis of molecular variance (AMOVA) among populations based on xvi cadre SSRs set.
TABLE S6 | Core cucumber varieties in Chinese market.
Footnotes
- ^ http://bigd.big.ac.cn/gsa
- ^ http://cucurbitgenomics.org/
- ^ http://pgrc.ipk-gatersleben.de/misa/
- ^ http://world wide web.fao.org/
References
Bo, M., Ma, Z., Chen, J., and Weng, Y. (2015). Molecular mapping reveals structural rearrangements and quantitative trait loci underlying traits with local accommodation in semi-wild Xishuangbanna cucumber (Cucumis sativus L. var. xishuangbannanesis Qi et Yuan). Theor. Appl. Genet. 128, 25–39. doi: x.1007/s00122-014-2410-z
PubMed Abstruse | CrossRef Full Text | Google Scholar
Botstein, D., White, R. 50., Skolnick, G., and Davis, R. W. (1980). Structure of a genetic linkage map in homo using brake fragment length polymorphisms. Am. J. Hum. Genet. 32, 314–331.
Google Scholar
Campbell, Due north. R., Harmon, S. A., and Narum, S. R. (2015). Genotyping-in-thousands past sequencing (GT-seq): a cost effective SNP genotyping method based on custom amplicon sequencing. Mol. Ecol. Resour. xv, 855–867. doi: ten.1111/1755-0998.12357
PubMed Abstract | CrossRef Full Text | Google Scholar
Costello, Thou., Fleharty, M., Abreu, J., Farjoun, Y., Ferriera, S., Holmes, L., et al. (2018). Label and remediation of sample index swaps by non-redundant dual indexing on massively parallel sequencing platforms. BMC Genomics nineteen:332. doi: 10.1186/s12864-018-4703-0
PubMed Abstruse | CrossRef Full Text | Google Scholar
Ding, S., Wang, S., Kang, H., Jiang, Chiliad., and Fei, Fifty. (2017). Large-scale analysis reveals that the genome features of uncomplicated sequence repeats are generally conserved at the family level in insects. BMC Genomics 18:848. doi: ten.1186/s12864-017-4234-0
PubMed Abstract | CrossRef Full Text | Google Scholar
Emanuelli, F., Lorenzi, Southward., Grzeskowiak, 50., Catalano, Five., Stefanini, M., Troggio, M., et al. (2013). Genetic diversity and population construction assessed by SSR and SNP markers in a large germplasm collection of grape. BMC Plant Biol. 13:39. doi: 10.1186/1471-2229-thirteen-39
PubMed Abstract | CrossRef Total Text | Google Scholar
Falush, D., Stephens, M., and Pritchard, J. K. (2003). Inference of population construction using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587.
PubMed Abstruse | Google Scholar
Gao, P., Ma, H., Luan, F., and Vocal, H. (2012). Dna fingerprinting of Chinese melon provides evidentiary support of seed quality appraisal. PLoS 1 7:e52431. doi: 10.1371/journal.pone.0052431
PubMed Abstract | CrossRef Full Text | Google Scholar
He, Ten., Li, Y., Pandey, Southward., Yandell, B. South., Pathak, M., and Weng, Y. (2013). QTL mapping of powdery mildew resistance in WI 2757 cucumber (Cucumis sativus Fifty.). Theor. Appl. Genet. 126, 2149–2161. doi: 10.1007/s00122-013-2125-6
PubMed Abstract | CrossRef Total Text | Google Scholar
Hu, J., Wang, P., Su, Y., Wang, R., Li, Q., and Sun, One thousand. (2014). Microsatellite diverseness, population structure, and core collection germination in melon germplasm. Establish Mol. Biol. Rep. 33, 439–447. doi: 10.1007/s11105-014-0757-6
CrossRef Total Text | Google Scholar
Huang, S., Li, R., Zhang, Z., Li, L., Gu, X., Fan, West., et al. (2009). The genome of the cucumber, Cucumis sativus 50. Nat. Genet. 41:1275. doi: x.1038/ng.475
PubMed Abstract | CrossRef Total Text | Google Scholar
Husson, F., Josse, J., Le, S., and Mazet, J. (2014). FactoMineR: Multivariate Exploratory Data Analysis and Information Mining with R. Boca Raton, FL: CRC Printing.
Google Scholar
International Union for the Protection of New Varieties of Plants [UPOV] (2011). Possible Used of Molecular Markers in the Examination of Distinctness, Uniformity and Stability (DUS). Geneva: UPOV.
Kamvar, Z. N., Tabima, J. F., and Grunwald, N. J. (2014). Poppr: an R package for genetic assay of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ two:e281. doi: 10.7717/peerj.281
CrossRef Total Text | Google Scholar
Lê, South., Josse, J., and Husson, F. (2008). FactoMineR: an R package for multivariate assay. J. Stat. Softw. 25, 1–eighteen.
Google Scholar
Li, Fifty., Fang, Z., Zhou, J., Chen, H., Hu, Z., Gao, L., et al. (2017). An accurate and efficient method for large-calibration SSR genotyping and applications. Nucleic Acids Res. 45:e88. doi: 10.1093/nar/gkx093
PubMed Abstract | CrossRef Full Text | Google Scholar
Li, Z., Zhang, Z., Yan, P., Huang, Due south., Fei, Z., and Lin, K. (2011). RNA-Seq improves notation of poly peptide-coding genes in the cucumber genome. BMC Genomics 12:540. doi: ten.1186/1471-2164-12-540
PubMed Abstract | CrossRef Full Text | Google Scholar
Lietzow, C. D., Zhu, H., Pandey, S., Havey, G. J., and Weng, Y. (2016). QTL mapping of parthenocarpic fruit set in North American processing cucumber. Theor. Appl. Genet. 129, ane–15.
PubMed Abstract | Google Scholar
Lu, H., Lin, T., Klein, J., Wang, S., Qi, J., Zhou, Q., et al. (2014). QTL-seq identifies an early on flowering QTL located well-nigh flowering locus T in cucumber. Theor. Appl. Genet. 127, 1491–1499. doi: 10.1007/s00122-014-2313-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Lv, J., Qi, J., Shi, Q., Shen, D., Zhang, S., Shao, Grand., et al. (2012). Genetic diversity and population structure of cucumber (Cucumis sativus L.). PLoS One 7:e46919. doi: 10.1371/journal.pone.0046919
PubMed Abstract | CrossRef Full Text | Google Scholar
Meynert, A. M., Ansari, M., Fitzpatrick, D. R., and Taylor, Grand. S. (2014). Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics 15:247. doi: x.1186/1471-2105-15-247
PubMed Abstract | CrossRef Full Text | Google Scholar
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590.
Google Scholar
Nishio, S. Y., Hayashi, Y., Watanabe, M., and Usami, S. I. (2015). Clinical application of a custom AmpliSeq library and ion torrent PGM sequencing to comprehensive mutation screening for deafness genes. Genet. Testing Mol. Biomarkers 19:209. doi: x.1089/gtmb.2014.0252
PubMed Abstract | CrossRef Full Text | Google Scholar
Pan, Y., Qu, S., Bo, Thou., Gao, M., Haider, G. R., and Weng, Y. (2017). QTL mapping of domestication and diversifying option related traits in circular-fruited semi-wild Xishuangbanna cucumber (Cucumis sativus L. var. xishuangbannanesis). Theor. Appl. Genet. 130, 1531–1548. doi: 10.1007/s00122-017-2908-2
PubMed Abstract | CrossRef Full Text | Google Scholar
Pritchard, J. One thousand., Stephens, Yard., and Donnelly, P. (2000). Inference of population construction using multilocus genotype data. Genetics 155, 945–959.
Google Scholar
Pritchard, J. K., Wen, X., and Falush, D. (2010). Documentation for Structure Software: Version ii.iii.
Google Scholar
Qi, J., Liu, X., Shen, D., Miao, H., Xie, B., Li, 10., et al. (2013). A genomic variation map provides insights into the genetic basis of cucumber domestication and multifariousness. Nat. Genet. 45, 1510–U1149. doi: x.1038/ng.2801
PubMed Abstract | CrossRef Full Text | Google Scholar
Song, Z. C., Miao, H., Zhang, S., Wang, Y., Zhang, Southward. P., and Gu, X. F. (2016). Genetic assay and QTL mapping of fruit peduncle length in cucumber (Cucumis sativus L.). PLoS One 11:e0167845. doi: ten.1371/journal.pone.0167845
PubMed Abstract | CrossRef Full Text | Google Scholar
Stewart, C. Northward. Jr., and Via, 50. East. (1993). A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. Biotechniques fourteen, 748–750.
PubMed Abstract | Google Scholar
Tan, J., Tao, Q., Niu, H., Zhang, Z., Li, D., Gong, Z., et al. (2015). A novel allele of monoecious (thousand) locus is responsible for elongated fruit shape and perfect flowers in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 128, 2483–2493. doi: 10.1007/s00122-015-2603-0
PubMed Abstract | CrossRef Full Text | Google Scholar
Tian, H. L., Wang, F. G., Zhao, J. R., Yi, H. M., Wang, Fifty., Wang, R., et al. (2015). Development of maizeSNP3072, a high-throughput compatible SNP assortment, for Deoxyribonucleic acid fingerprinting identification of Chinese maize varieties. Mol. Breed. 35:136. doi: 10.1007/s11032-015-0335-0
PubMed Abstract | CrossRef Total Text | Google Scholar
Tóth, G., Gáspári, Z., and Jurka, J. (2000). Microsatellites in dissimilar eukaryotic genomes: survey and assay. Genome Res. 10:967. doi: 10.1101/gr.10.7.967
CrossRef Full Text | Google Scholar
Vähä, J. P., Erkinaro, J., Niemelä, E., and Primmer, C. R. (2007). Life-history and habitat features influence the inside-river genetic structure of atlantic salmon. Mol. Ecol. 16, 2638–2654. doi: 10.1111/j.1365-294x.2007.03329.10
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, F., Yang, Y., Yi, H., Zhao, J., Ren, J., Wang, L., et al. (2017). Construction of an SSR-based standard fingerprint database for corn multifariousness authorized in China. Sci. Agric. Sin. 50, 1–14.
Google Scholar
Wang, L. Ten., Qiu, J., Chang, L. F., Liu, L. H., Hong-Bo, L. I., Pang, B. S., et al. (2015). Cess of wheat variety distinctness using SSR markers. J. Integr. Agric. 14, 1923–1935. doi: 10.1016/s2095-3119(15)61057-7
CrossRef Full Text | Google Scholar
Wang, 10., and Le, W. (2016). GMATA: an integrated software package for genome-calibration SSR mining, marker development and viewing. Forepart. Found Sci. seven:1350. doi: 10.3389/fpls.2016.01350
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, Y., Vandenlangenberg, One thousand., Wehner, T. C., Kraan, P. A. 1000., Suelmann, J., Zheng, X., et al. (2016). QTL mapping for featherlike mildew resistance in cucumber inbred line WI7120 (PI 330628). Theor. Appl. Genet. 129, one–13. doi: 10.1007/s00122-016-2719-ten
PubMed Abstract | CrossRef Full Text | Google Scholar
Weng, Y., Colle, G., Wang, Y., Yang, 50., Rubinstein, Thousand., Sherman, A., et al. (2015). QTL mapping in multiple populations and development stages reveals dynamic quantitative trait loci for fruit size in cucumbers of different market classes. Theor. Appl. Genet. 128, 1747–1763. doi: 10.1007/s00122-015-2544-seven
PubMed Abstract | CrossRef Full Text | Google Scholar
Wright, Southward. (1965). The interpretation of population structure by F-statistics with special regard to systems of mating. Development 19, 395–420. doi: 10.1111/j.1558-5646.1965.tb01731.x
CrossRef Full Text | Google Scholar
Wu, Z., Zhang, T., Li, L., Xu, J., Qin, X., Zhang, T., et al. (2016). Identification of a stable major-effect QTL (Parth 2.1) decision-making parthenocarpy in cucumber and associated candidate gene analysis via whole genome re-sequencing. BMC Found Biol. 16:182. doi: 10.1186/s12870-016-0873-six
PubMed Abstract | CrossRef Full Text | Google Scholar
Xu, X., Jing, J., Qiang, Ten., Qi, Ten., and Chen, X. (2017). Inheritance and quantitative trail loci mapping of accidental root numbers in cucumber seedlings under waterlogging conditions. Mol. Genet. Genom. 292, 353–364. doi: 10.1007/s00438-016-1280-2
PubMed Abstract | CrossRef Full Text | Google Scholar
Yang, J., Zhang, C., Zhao, North., Zhang, L., Hu, Z., Chen, Due south., et al. (2017). Chinese root-type mustard provides phylogenomic insights into the evolution of the multi-utilise diversified allopolyploid Brassica juncea. Zeitschrift Fur Gastroenterologie 152:S695.
PubMed Abstruse | Google Scholar
Yang, S., Fresnedoramírez, J., Wang, Chiliad., Cote, L., Schweitzer, P., Barba, P., et al. (2016). A next-generation mark genotyping platform (AmpSeq) in heterozygous crops: a case report for marker-assisted selection in grapevine. Hortic. Res. 3:16002. doi: 10.1038/hortres.2016.two
PubMed Abstruse | CrossRef Full Text | Google Scholar
Yasodha, R., Vasudeva, R., Swati, B., Sakthi, A. R., Abel, N., Binai, N., et al. (2018). Draft genome of a high value tropical timber tree, Teak (Tectona grandis L. f): insights into SSR diverseness, phylogeny and conservation. Deoxyribonucleic acid Res. 25, 409–419. doi: ten.1093/dnares/dsy013
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhang, H., Fan, J., Guo, South., Ren, Y., Gong, G., Zhang, J., et al. (2016). Genetic diversity, population structure, and formation of a core collection of 1197 citrullus accessions. Hortscience 51, 23–29. doi: 10.21273/hortsci.51.1.23
CrossRef Full Text | Google Scholar
Zhang, H., Wang, H., Guo, South., Ren, Y., Gong, G., Weng, Y., et al. (2012). Identification and validation of a core set of microsatellite markers for genetic diversity analysis in watermelon, Citrullus lanatus thunb. matsum. & nakai. Euphytica 186, 329–342. doi: 10.1007/s10681-011-0574-z
CrossRef Full Text | Google Scholar
Source: https://www.frontiersin.org/articles/450617
0 Response to "How to Read an Ssr Pedigree Tree"
Post a Comment